本帖最后由 costa_na 于 2014-7-26 13:01 编辑
先搬过来再翻译
Will intermittent dosing transform TKI therapy in cancer research?
间歇性用药会改变TKI治疗在肿瘤研究中应用(的现状)吗?
At AACR last year, one of the most revealing presentations was on metastatic melanoma, specifically, some elegant research by Meghna Das Thakur (NIBR) demonstrating that intermittent pulsing of vemurafenib (a BRAF V600E inhibitor) led to less resistance than inhibiting the target 24/7.
去年的AACR上,在那些最具启发意义的报告中,有一个是由Meghna Das Thakur阐述的关于转移性黑色素瘤的一些很漂亮的研究,其展示了对比不间断用药,vemurafenib(一种BRAF V600E抑制剂)的间歇性脉冲用药导致了更低程度的耐药。
Many of us wondered whether such a pulsing approach would be useful for other tyrosine kinase inhibitors (TKIs).
我们许多人都想知道类似的脉冲方案是否能够应用于其他的酪氨酸激酶抑制剂(TKIs)
Fast forward to this week.
(话题)转到这周来*。
Neal Rosen’s lab at MSKCC has an interesting new paper out looking at the effects of pulse dosing with PI3K and ERK inhibition, since targeting both has long been suspected to be key in overcoming cross-resistance.
纪念斯隆-凯特琳癌症中心(MSKCC)的Neal Rosen实验室发表了一篇有意思的新论文,其研究了PI3K和ERK抑制剂脉冲给药的有效性,因为长久以来一直都认为同时靶向这两者是克服交叉耐药的关键所在。
Recall that despite promising preclinical research, most of the early patient trials looking at targeting the PI3K-Akt-mTOR and RAS-RAF-MEK-ERK pathways in combination were, however, disappointing to say the least, both in terms of the side effect profile, and also with respect to clinical efficacy.
回想起尽管有一些有希望的临床前研究,但不夸张地说,绝大多数关于联合靶向PI3K-Akt-mTOR和RAS-RAF-MEK-ERK信号通路的早期临床研究,在副作用和临床有效性这两方面,都是令人失望的。
These results also applied to combinations with chemotherapy, which were added to either agent to try and induce cell death via apoptosis.
将其中一种药物增加到化疗中试图借助凋亡引导细胞死亡,这种与化疗的联合也是同样的(令人失望的)结果。
We know that the PI3K pathway is dysregulated in many cancers, so why have the combinations tried to date produced less than optimal results?
我们知道,在很多肿瘤中都存在PI3K通路的失调,所以为什么这些联合(用药)没有达到最理想的效果呢?
Well, Will et al., (2014) showed that:
- RAS-ERK pathway is a key downstream effector pathway of oncogenic PI3K
- ERK inhibition is required for apoptosis (cell death) to occur with a PI3K inhibitor
- It is important to coordinate downregulation of AKT and ERK since both are necessary for induction of apoptosis and antitumor activity
- Such an effect can be achieved with intermittent dosing, which will also likely decrease toxicity and allow administration of therapeutic doses
Will在今年的一篇论文中写道:
- RAS-ERK通路是致瘤性PI3K的一个关键的下游效应器通路
- 在使用PI3K抑制剂的同时抑制ERK,对于凋亡(细胞死亡)来说是必要的。
- 因为(抑制)AKT和ERK对于诱导凋亡和(药物的)抗肿瘤活性来说是必须的,所以重要的是协同下调这两者
- 类似的效果可以通过间歇性脉冲(给药)来实现,这样可能会降低毒性同时也可用控制好治疗的剂量
Ah so the same concept that Das explored in metastatic melanoma could also have potential with PI3K and MEK inhibition!
所以Das在转移性黑色素瘤中所探索的相同的概念,在对PI3K和MEK抑制作用上也有应用的潜力。
I find this approach fascinating because in the past, when I queried whether we needed to hit two targets maximally and continuously, rather than look at intermittent or minimally effective dosing (MED), industry people were up in arms and sent me more heated emails on this topic than anything else we’ve ever blogged about!
我个人觉得这种方案棒极了,因为在以前当我询问我们能否(采用)最大剂量且持续地攻击两个靶点,而不是间歇性地或者以最低有效剂量(来给药)的时候,药厂的人们表示极力反对并且发送了比这个博客里任何一篇文章都要更愤怒的邮件给我!
Meanwhile, Rosen himself hinted at this solution in a talk at the AACR Molecular Targets meeting in Boston last year and said a publication was underway to explain their findings. Generally, I don’t report on unpublished findings out of respect to the scientists and thus didn’t mention it in our extensive AACR Targets Coverage, but am delighted this is now a topic for more public discussion.
同时,Rosen自己在去年波士顿召开的AACR分子靶向会议的一次谈话中也间接提到了这种方案,那时他就说(他的研究小组)正在撰写一篇论文来解释他们的发现。一般来说,出于对科学家们的尊敬,我不会报告那些未公开发表的发现,所以我并没有在我们的AACR靶向全面报道中提到此事,但现在我很高兴因为有更多的关于这个话题的公开讨论了。
Part of the conundrum was articulated by Will et al., (2014) in their author manauscript (see below for the link under the Cancer Discovery Online First Section this month):
这个难题的一部分在Will et al., (2014)的作者手稿中得到了清晰地阐述(参见下面的链接,发表于这个月的Cancer Discovery First Section):
Since mTOR and AKT inhibitors reactivate PI3K signaling, we asked whether PI3K inhibitors have more significant antitumor activity, perhaps by inhibiting other PI3K targets in addition to AKT/mTOR.
因为mTOR和AKT抑制剂重新激活了PI3K信号,我们想到是否PI3K抑制剂具有更显著的抗肿瘤活性,也许这能通过抑制其他PI3K靶点联合抑制AKR/mTOR(来实现)。
Selective PI3K and AKT inhibitors were compared in tumors with activation of PI3K pathway signaling in order to assess differences in the biochemical and biologic consequences of their inhibition. Both inhibitors effectively inhibited downstream targets of AKT, relieved feedback inhibition of growth factor receptors, and inhibited cell growth. However, in HER2-dependent breast cancers, PI3K inhibitors, but not AKT inhibitors, caused the rapid induction of a significant degree of apoptosis.
选择性的PI3K和AKT抑制剂在具有激活的PI3K信号通路的肿瘤中进行了对比,以评估两者在生化特性和抑制作用的生物学效果方面的差异。两种抑制剂都能有效抑制AKT下游靶点,这减弱了生长因子受体(growth factor receptors)的反馈抑制,从而抑制了细胞生长。但是在HER2依赖的乳腺肿瘤中,PI3K抑制剂,而不是AKT抑制剂,引起了对极高程度凋亡的快速诱导作用。
We find that, whereas AKT inhibitors inhibit AKT/mTOR and activate ERK signaling, PI3K inhibitors inhibit both. They cause durable inhibition of AKT signaling but also transient inhibition of RAS activation and ERK signaling, both of which are required for induction of apoptosis. Moreover, induction of apoptosis by an AKT inhibitor is significantly enhanced when combined with a MEK inhibitor.
我们发现,尽管AKT抑制剂抑制了AKT/mTOR却激活了ERK信号,但PI3K抑制剂能同时抑制这两者。PI3K抑制剂能够导致持续的AKT信号抑制作用,但同时却(只能)短暂地抑制RAS活化以及ERK信号,这两者(的抑制)都是诱导凋亡所必须的。进一步说,通过一种AKT抑制剂来诱导凋亡,这种效应可以由联合一种MEK抑制剂得到显著的增强。
Our results show that PI3K is upstream of wild type RAS as well as AKT/mTOR, and this causes the therapeutic consequences of PI3K inhibition to be significantly greater than those of AKT inhibition.
我们的研究结果显示,PI3K是野生型RAS和AKT/mTOR的上游通路,这可以导致抑制PI3K(所产生)的治疗效果要显著好于对AKT的抑制。
A number of different inhibitors of PI3K, AKT, mTOR and MEK were explored in this research, so the results are not limited to one or two.
这个研究分析了许多不同的PI3K、AKT、mTOR和MEK抑制剂,所以其结果并不仅限于一两种。
One important question that the group sought to address the inhibition issue:
该小组寻求解决抑制难题(所面临的)的一个重要问题是:
PI3K inhibitors cause rapid inhibition of ERK in breast cancer cells with HER2 amplification, but P-ERK levels rebound fairly quickly. Even so, this transient inhibition is required for significant induction of apoptosis by these drugs. We asked whether more complete and sustained inhibition of ERK might enhance induction of cell death by the PI3K inhibitor.
PI3K抑制剂在具有HER2扩增的乳腺癌细胞中导致对ERK的快速抑制。但ERK的磷酸化水平(P-ERK level)反弹地极其迅速。即便如此,这种短暂的抑制对于采用这类药所诱导的显著的细胞凋亡来说是必须的。我们想到是否能够通过PI3K抑制剂联合对ERK更全面和持久的抑制,从而增强对肿瘤细胞死亡的诱导作用。
They found that:
他们发现:
These results suggest that, in some cells, inhibition of other non-AKT targets of PI3K contribute to induction of apoptosis, or that stronger MEK inhibition is required to fully activate apoptosis. Combined inhibition of MEK and PI3K caused more apoptosis than any of the other treatments in all three models.
这些结果表明,在某些细胞中,对其他非AKT靶点的PI3K的抑制有助于诱导凋亡,或者全面的活化凋亡需要对MEK更强力的抑制。在这三种模型中,联合抑制MEK和PI3K能产生比其他治疗方案更高程度的促凋亡(效果)。
This lead to further work and the finding that:
这产生了更进一步的工作,以及(下面的研究)结果:
Pulsatile PI3K inhibition caused initial tumor regression and significantly suppressed tumor growth. The effectiveness of intermittent administration of the PI3K inhibitor and its superior antitumor activity compared to AKT inhibition were confirmed in another HER2 amplified, PI3K mutant breast cancer model, MDA-MB–361.
对PI3K的脉冲抑制在最开始就导致了肿瘤的缩小和对肿瘤生长的显著抑制。PI3K抑制剂间歇性给药的有效性,以及其相对AKT抑制剂更好的抗肿瘤活性在另一种具有HER2扩增和PI3K突变的乳腺癌肿瘤模型,MDA-MB-361,中得到了确认。
The reason for this?
其原因是什么呢?
We hypothesized that the effectiveness of PI3K inhibition was due in part to its combined inhibition of ERK and AKT.
我们假定抑制PI3K的有效性,部分是由于其联合抑制了ERK和AKT。
The Will et al., (2014) article is available online and open access (see here for direct link) – I highly recommend those interested in this field checking it out and reading the nuggets for yourself, it’s well written and easy to follow.
Will的文章可以在线获取和访问(看下面的链接),我强烈推荐对这个领域有兴趣的同仁下载下来仔细阅读,并且从里挖出对你自己有价值的东西,这篇文章写得很棒并且很容易理解。
What does all this mean?
全部这些说明了什么呢?
It would be hard for me to improve on Will et al., (2014) conclusion that:
对我来说,Will文章里的概括是最好的(回答):
Recently, treatment with more selective PI3K inhibitors has led to greater therapeutic efficacy in lymphomas and in breast cancer with PI3K mutation or HER2 amplification. The ability of any PI3K inhibitor to inhibit signaling adequately is limited by physiologic toxicity. Moreover, attempts to combine MEK inhibitors with `dual specificity’ PI3K or AKT inhibitors have been complicated by severe toxicity at modest doses of these drugs.
The idea that the pathway must be inhibited continuously dominates the clinical development of these drugs.
Our finding that transient inhibition of PI3K is effective in in vivo models suggests that periodic rather than continuous target inhibition is an alternative strategy that would allow adequate pathway inhibition without causing inordinate toxicity or chronic feedback reactivation of receptors.
Thus, combining PI3K inhibitors, MEK inhibitors and, perhaps, inhibitors of key reactivated RTKs, and administering them at high dose on intermittent schedules may be a more effective therapeutic strategy for these tumors.
Overall, don’t be surprised to suddenly see new clinical trials emerge evaluating intermittent dosing with PI3K and MEK inhibitors. The only questions in my mind is who will be the first to go this route and who will be able demonstrate superior efficacy and tolerability in patients?
The scientific rationale is very solid for intermittent dosing with BRAF V600E inhibitors and now with the combination with a PI3K plus a MEK inhibitor; it will be really interesting to see if such an approach will translate successfully in the clinic. I hope it does because improving outcomes is
ultimately what we are all here for.
*: 该博文发表于2014年1月17日 |
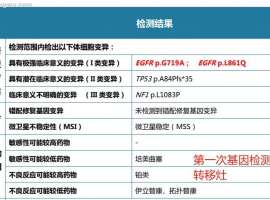 G719A+L861Q双罕见突变治疗24个月,
2022.12 胸闷咳嗽,肺CT发现3.1*2.8占位,双肺多发微小结节,增强CT占位有增强,胸椎3
G719A+L861Q双罕见突变治疗24个月,
2022.12 胸闷咳嗽,肺CT发现3.1*2.8占位,双肺多发微小结节,增强CT占位有增强,胸椎3
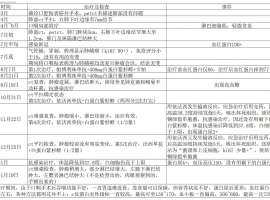 依沃西用药后,该怎么办?
依沃西用药后,该怎么办?
我爸爸于2024年7月确诊肺鳞癌,整个治疗过程如附件里表格
依沃西用药后,该怎么办?
依沃西用药后,该怎么办?
我爸爸于2024年7月确诊肺鳞癌,整个治疗过程如附件里表格
 pet-ct结果,请大佬们受累给看一下。
节后在心胸外科住院,马上安排了pet-ct,今天拿到报告,请大佬们受累给解读一下,结节
pet-ct结果,请大佬们受累给看一下。
节后在心胸外科住院,马上安排了pet-ct,今天拿到报告,请大佬们受累给解读一下,结节
 求助 K药和替雷利珠如何选择
父亲9月刚查出非小细胞低分化癌伴随左肾上腺和第10胸椎转移PDL1高表达(TPS=90%) SMARC
求助 K药和替雷利珠如何选择
父亲9月刚查出非小细胞低分化癌伴随左肾上腺和第10胸椎转移PDL1高表达(TPS=90%) SMARC
 需要加论坛患者群的朋友们看这里
本帖最后由 青菜567 于 2024-7-22 17:38 编辑
与癌共舞小助手-29新进论坛需要进入患
需要加论坛患者群的朋友们看这里
本帖最后由 青菜567 于 2024-7-22 17:38 编辑
与癌共舞小助手-29新进论坛需要进入患




 显身卡
显身卡





